Rationale & Aims - Rare Cancers Genomics
RATIONALE
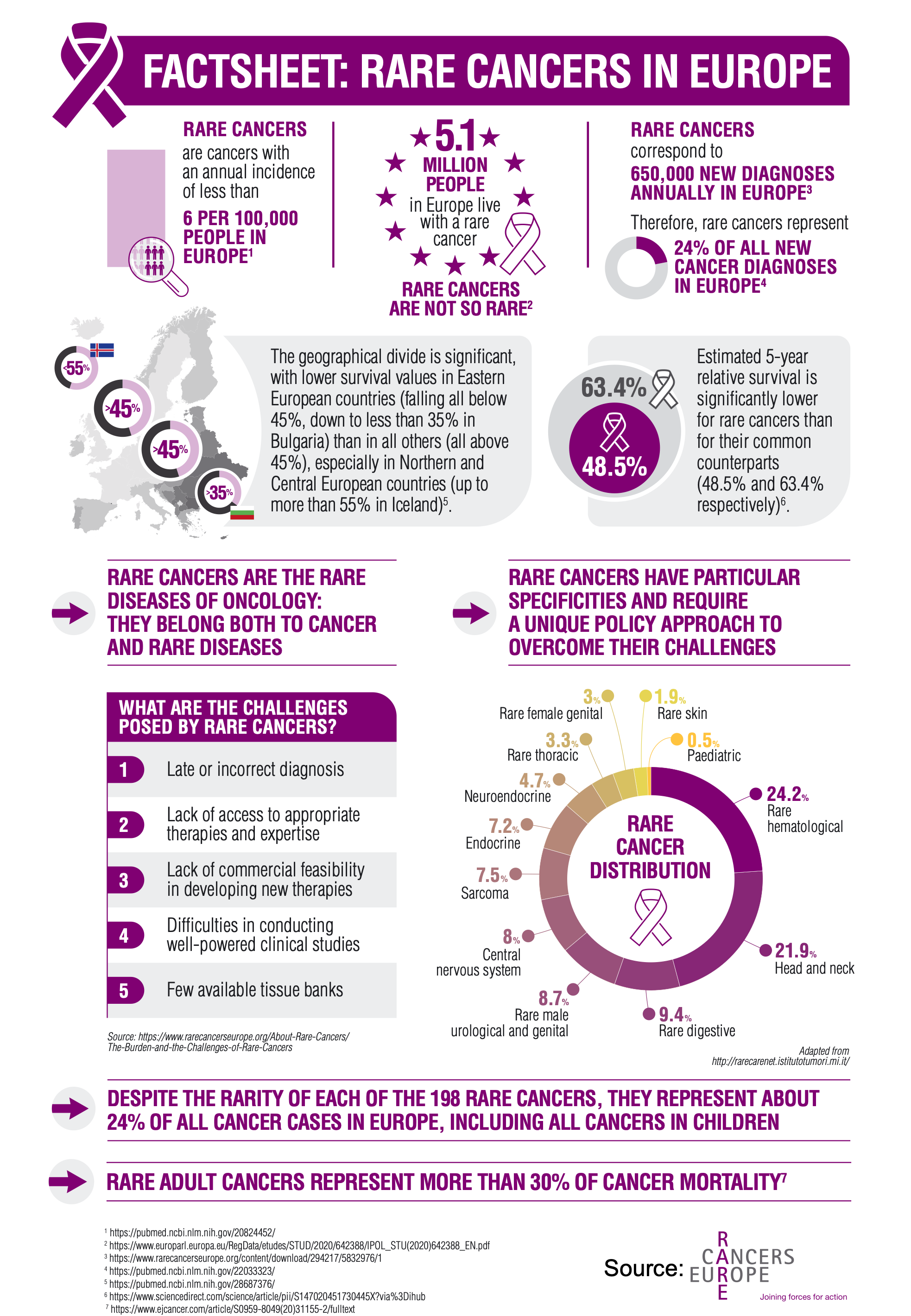
Rare cancers (<6/100,000) account for 25% of cancer deaths, representing a substantial burden of disease (Greenlee, 2010). Twelve families for a total of 198 rare cancers have been identified by the RARECARE project, based on the International Classification of Diseases for Oncology (ICD-O, 2013): central nervous system, digestive, endocrine organ, female genital, hematological, head and neck, male genital and urogenital, neuroendocrine tumors, pediatric cancers, sarcomas, skin, and thoracic (including malignant mesothelioma). Although individually rare, collectively, rare cancers represent about 24% of all cancer cases diagnosed each year (Gatta, 2011; Gatta, 2017). While tens of thousands of patients may be living with a given rare cancer, their geographic dispersion often limits the number of cases seen at any one institution. The fewer experts translate into a need for centralized patient referral and, as a result, delays in obtaining a correct diagnosis. Low incidence rates also make clinical trial recruitment for rare cancers extremely challenging, stalling efforts to test emerging therapeutic hypotheses (Sharifnia, 2017). The limited amount of data available affects the feasibility of developing models that meet good practice recommendations for health technology assessment submissions (Pearson, 2018). Therefore, lack of treatment guidelines, fewer clinical trials dedicated to rare cancers, and less investment in drug development are factors contributing to poor outcomes (Blay, 2016; Panageas, 2015).
As a result, rare cancer patients have worse outcomes, with estimated average 5-year relative survival rates of 47% versus 65% for those with common cancers (Gatta, 2011). In basic research settings, a scarcity of rare tumor tissue and patient-derived models can preclude well-powered studies aimed at elucidating the underlying biology (Boehm, 2015). Overall, basic biological and clinical knowledge for rare cancers is lacking: rare cancers are understudied, economies of scale cannot be made, there is not a large enough market to incentivize drug development, and benefits in outcomes for new therapeutic strategies cannot be demonstrated through conventional studies (Rare Cancers Europe). Many of the barriers to research directly stem from the low incidence rates of individual tumor types. Identifying new approaches to study rare cancers that help overcome the intrinsic limitation of low numbers would improve the clinical management and health outcomes of these discriminated patients.
AIMS
The Rare Cancers Genomics initiative is an international multidisciplinary open-science effort to shed light on the molecular characteristics of rare cancers, to understand their etiology and carcinogenesis processes, and to ultimately improve their clinical management and consequently, patient’s prognosis.
RESEARCH APPROACH
To achieve our aims we follow different approaches:
1
Perform integrative multi-omics multi-region bulk and single-cell sequencing analyses as well as spatial profiling of large bio-repositories with good quality of samples and detailed pathological, clinical and epidemiological annotations.
(led by Drs Lynnette Fernandez-Cuesta and Matthieu Foll)
2
Review and identify new morphological characteristics using image-based AI and integrate them with the molecular data.
(led by Drs Matthieu Foll and Lynnette Fernandez-Cuesta – read more)
3
Study cancer ecology and evolution to identify and understand carcinogenic processes.
(led by Dr Nicolas Alcala – read more)
4
Use state-of-the-art in vitro organoid models to study the cancer initiation and progression.
(led by Dr Talya Dayton – read more)
OPEN SCIENCE EFFORT
This initiative includes a strong commitment to open science, reproducibility, and capacity building: best-practices pipelines have been set-up to analyze WGS, RNAseq, and methylation data, as well as supervised and unsupervised methods to perform multi-omic data integration. All the necessary resources to reproduce the initiative’s analyses are available, and the bioinformatics pipelines are continuously updated and improved, with detailed documentation to ensure that they are reproducible and effectively reusable by others. Similar single-cell RNA- seq, ATAC-seq, and spatial RNA-seq data processing workflows following best practices (Andrews, 2021) are currently being developed.
