Bioinformatics
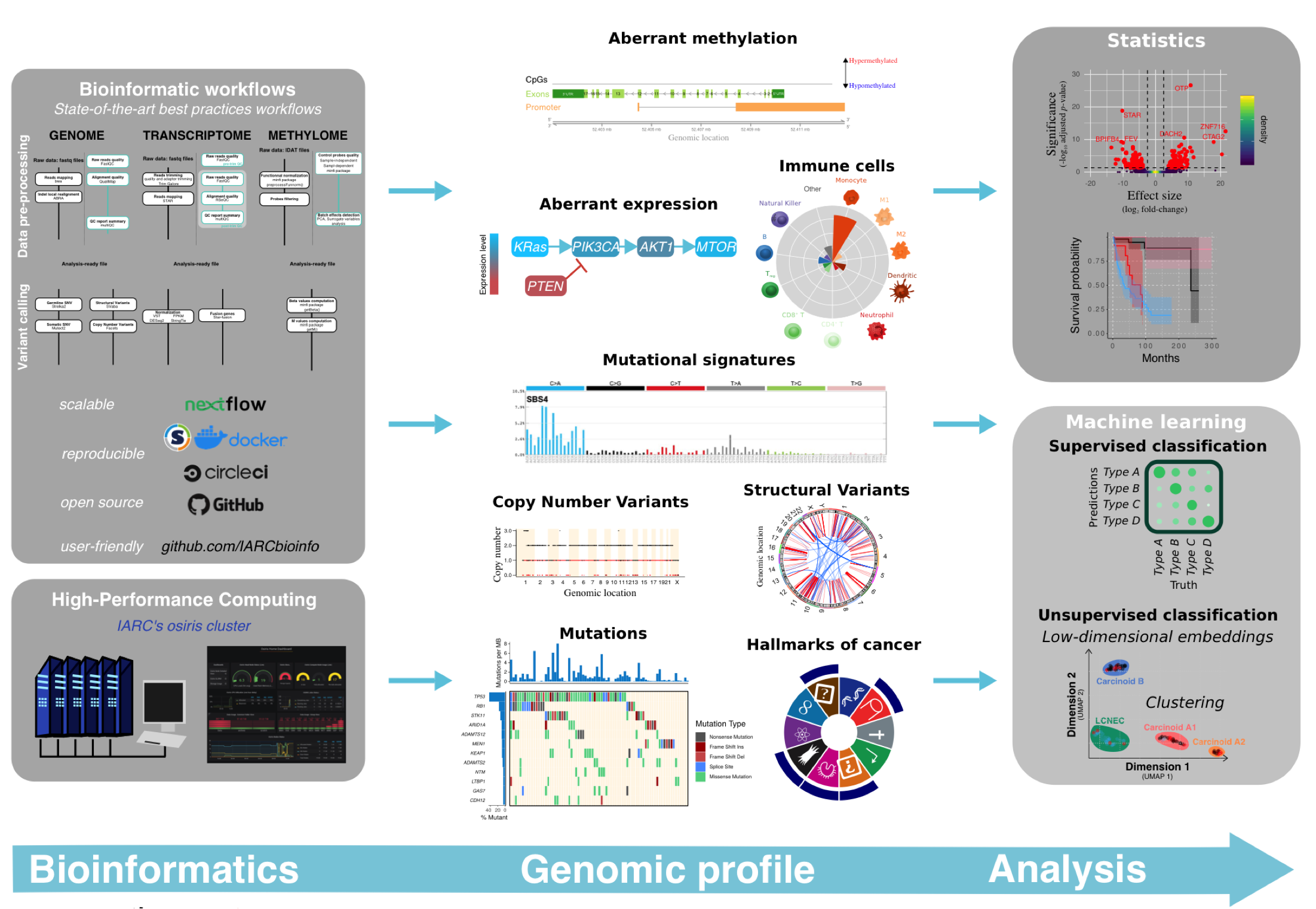
Our approach consists in using state of the art computational methods to analyse omics data. We are very committed to open science and therefore implement, document, and release as open-source projects to the community everything we produce. Our tools rely on the most recent informatics technologies (GitHub, (Bio)conda, Docker, Singularity, circleCI, Nextflow) to ensure a perfect reproducibility of results, scalability–from individual computers to large high-performance computing facilities–, and portability to suit a maximal range of platforms (Di Tommaso et al. Nature Biotechnology 2017). All our pipelines are available at https://github.com/IARCbioinfo.
When we design bioinformatics pipelines we adhere to the KISS principle (« keep it simple, stupid »): most systems work best if they are kept simple. For this reason we try to have pipelines that do only one task (e.g. DNA sequencing alignment) but do it well rather than having a single « genomics pipeline » that would do everything automatically but that would be hard to read, to maintain, and not modular.
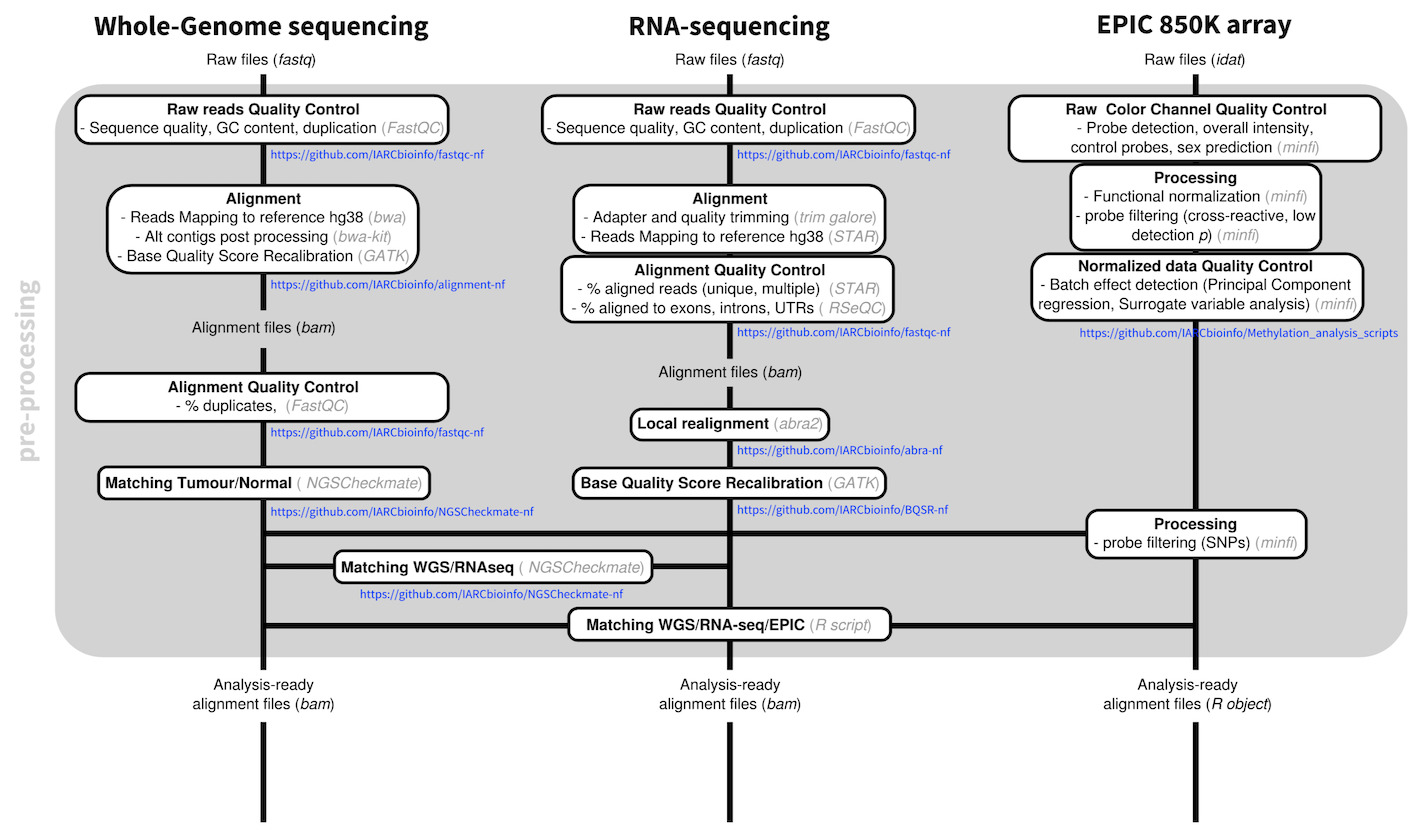
We have set up best-practices pipelines to pre-process WGS, RNA-seq, and methylation array data. These pipelines include pre-processing steps for each data type (e.g., sequence alignment and base quality score recalibration, array signal normalization) and rigorous quality control at each step (e.g., sequence quality, GC content, duplication, array control probes).
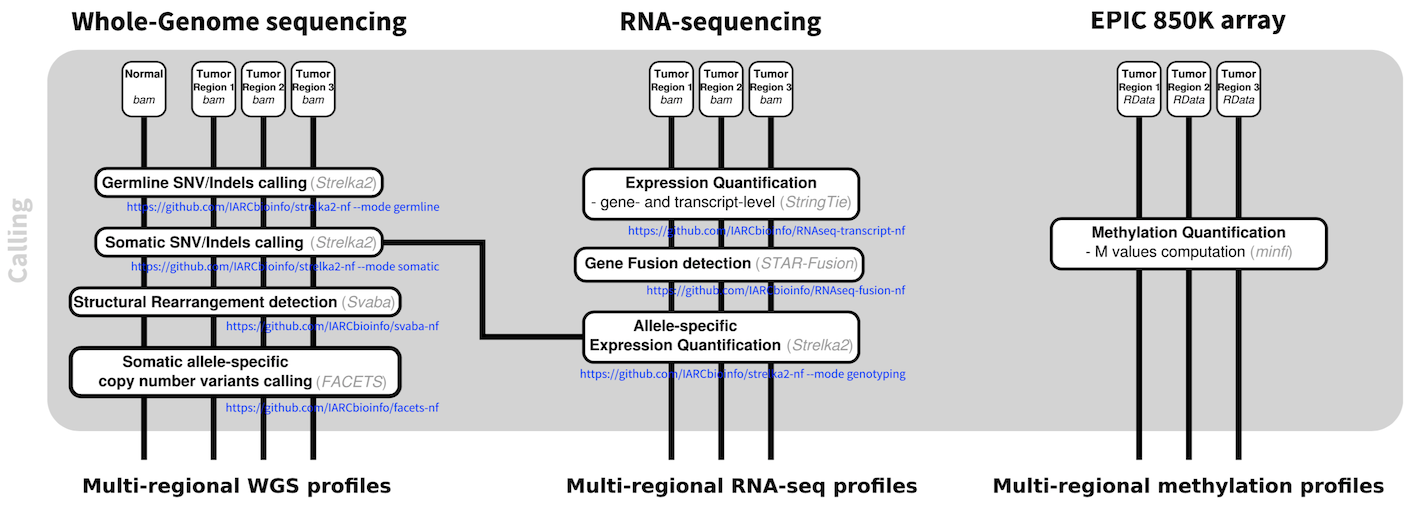
Furthermore, we have set up the analysis pipelines to call single nucleotide variants and indels, copy number variants, genomic rearrangements, gene fusions, and to perform gene and transcript-level expression quantification and methylation quantification.
Finally, we have also developed the supervised and unsupervised methods to perform integration of multi-omic datasets (also available at https://github.com/IARCbioinfo).