
SCIENTIFIC HIGHLIGHTS
It is widely accepted that lung neuroendocrine tumours (NETs) and lung neuroendocrine carcinomas (NECs) are different diseases and not simply a continuum of neoplasms with a common pathogenesis. However, over the past years it has been suggested that in both the lung and the thymus the progression or transition from NET to NEC, possibly through the accumulation of genetic anomalies, might be possible. In line with this hypothesis and through multi-omics factor analysis of 116 pulmonary carcinoids (including 35 atypical), 75 large-cell neuroendocrine carcinomas (LCNEC), and 66 small-cell lung cancers, we have identified a group of atypical carcinoids that we have named “supra-carcinoids” with carcinoid-like morphology yet the molecular and clinical features of the deadly LCNEC, suggesting that the molecular link between lung NETs and NECs might be subtler than initially thought (Figure below). In the same study we identified clinically relevant molecular groups of pulmonary carcinoids (Alcala et al., Nat Commun 2019).
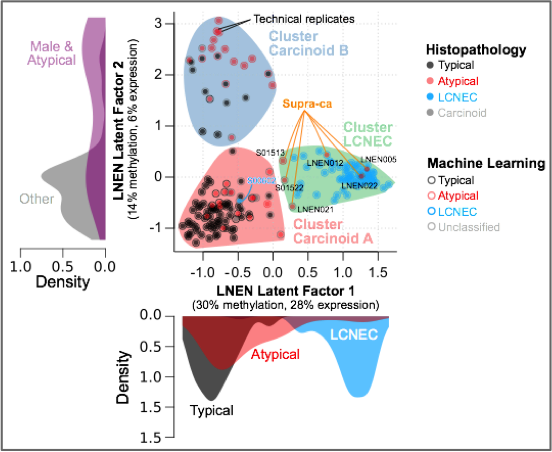
Multi-omics factor analysis (MOFA) of transcriptomes and methylomes of LNEN samples (typical carcinoids, atypical carcinoids, and LCNEC). Point colours correspond to the histopathological types; coloured circles correspond to predictions of histopathological types by a machine learning (ML) algorithm (random forest classifier); filled coloured shapes represent the three molecular clusters identified by consensus clustering. The density of clinical variables that are significantly associated with a latent factor (ANOVA q-value < 0.05) are represented by kernel density plots next to each axis: histopathological type for latent factor 1, sex and histopathological type for latent factor 2.
Another important contribution that we would like to highlight is the generation of a molecular map of lung neuroendocrine neoplasms (Figure below), which provides an interactive way to explore the molecular data and allows easy statistical interrogation, including generating new hypotheses, but also projecting data from studies including fewer samples, (so frequent when working with rare cancers) in order to draw meaningful conclusions (Gabriel et al. Gigascience 2020).
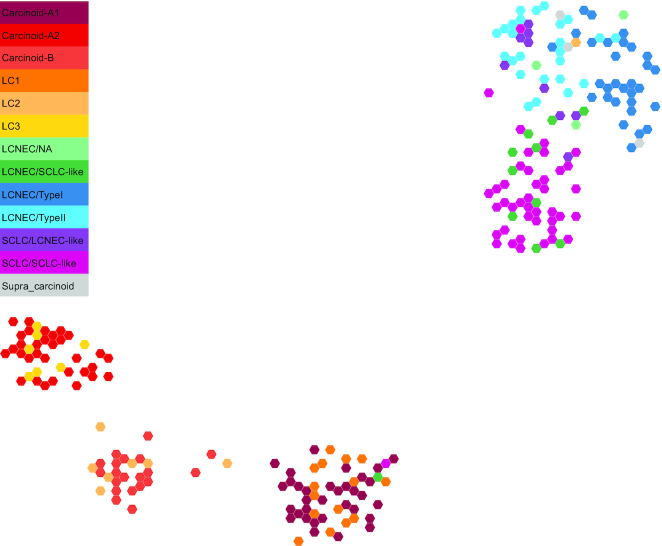
Two-dimensional projection of LNEN transcriptome data using UMAP. The representation was obtained from the TumorMap portal, using the hexagonal grid view, each hexagonal point representing a LNEN sample. Point colours correspond to the molecular clusters defined in the previous publications.
We are currently performing a full molecular characterization of atypical carcinoids and supra-carcinoids (an aggressive group of pulmonary carcinoids that we have recently identified) through a multi-omic integrative analysis (WGS, RNA-seq, and 850k methylation arrays) of 100 primary tumors. The molecular data will be correlated with morphological, epidemiological, and clinical features. Finally, together with T Dayton (in Clever’s lab) we are trying to depict the mechanisms of a possible progression from low to high-aggressive lung neuroendocrine neoplasms by modeling tumor initiation and progression, using state-of-the-art organoid in vitro models.
VIDEOS
FUNDING
Filling the unexpected gap from supra-carcinoids to large-cell neuroendocrine carcinomas: shedding light on the genesis of high-grade lung neuroendocrine neoplasms. Worldwide Cancer Research (WCR, UK). 2025 Grant Round. Coordinators. Active
Characterization of supra-carcinoid cell states to inform interception strategies. Neuroendocrine Tumor Research Foundation (NETRF, US). 2025 Petersen Accelerator Award. Coordinators. Active
Reconstructing the evolutionary history of neuroendocrine tumor subtypes. Neuroendocrine Tumor Research Foundation (NETRF, US). 2023 Mentored Award. Coordinators. Completed
Reconciling lung carcinoids histopathological and molecular classifications. Neuroendocrine Tumor Research Foundation (NETRF, US). 2022 Investigator Award. Coordinators. Completed – IARC Press Release – NETRF 2022 Annual Report
Unveiling the evolutionary processes and molecular pathways underlying the development and progression of lung neuroendocrine neoplasms. Worldwide Cancer Research (WCR, UK). 2020 Grant Round. Coordinators. Completed
Comprehensive molecular characterisation of lung supra-carcinoids. Neuroendocrine Tumor Research Foundation (NETRF, US). 2019 Investigator Award. Coordinators. Completed
Genomic characterisation of broncho-pulmonary carcinoids. Institut National Du Cancer (INCa, France). PRT-K17-047. Coordinators. Completed
The Orthopedia Homeobox transcription factor (OTP) in the diagnostics and tumorigenesis of lung carcinoids. KWF Kankerbestrijding (DCS, The Netherlands). Collaborators. Completed
Epigenomic characterisation of lung neuroendocrine tumours. Ligue Contre le Cancer (LNCC, France). Coordinators. Completed
Genomic and transcriptomic characterisation of atypical carcinoids of the lung. National Institutes of Health (NIH, US). R03 CA195253-01. Coordinators. Completed


SCIENTIFIC HIGHLIGHTS
Molecular and immunologic breakthroughs are transforming the management of thoracic cancer, although advances have not been as marked for malignant pleural mesothelioma where pathologic diagnosis has been essentially limited to three histologic subtypes. In order to go towards a more multidisciplinary approach for the histologic classification of MPM, we participated in a multidisciplinary group (pathologists, molecular biologists, surgeons, radiologists, and oncologists), sponsored by EURACAN/IASLC to critically review the current classification and issue recommendations. These multidisciplinary recommendations for pathology classification and application will allow more informative pathologic reporting and potential risk stratification, to support clinical practice, research investigation and clinical trials.
While generating the multi-omic data for our own series of MPMs, we started by reanalysing the publicly available data (Bueno et al. Nat Genet. 2016; TCGA Cancer Discov. 2019) with no assumption of discreteness, i.e., without assuming the existence of groups, which is the way the other studies analysed the data. This idea came from the fact that already at the histological level a continuum is suggested, with biphasic MPMs having from 10 to 90% or epithelioid and sarcomatoid components. For this, we undertook unsupervised analyses of RNA-sequencing data of 284 MPMs, and we identified a continuum of molecular profiles that explained the prognosis of the disease better than any discrete model. The immune and vascular pathways were the major sources of molecular variation, with strong differences in the expression of immune checkpoints and pro-angiogenic genes; the extrema of this continuum had specific molecular profiles: a “hot” bad-prognosis profile, with high lymphocyte infiltration and high expression of immune checkpoints and pro-angiogenic genes; a “cold” bad-prognosis profile, with low lymphocyte infiltration and high expression of pro-angiogenic genes; and a “VEGFR2+/VISTA+” better-prognosis profile, with high expression of immune checkpoint VISTA and pro-angiogenic gene VEGFR2 (Figure below). We validated the gene expression levels at the protein level for a subset of five selected genes belonging to the immune and vascular pathways, in a subset of 103 samples and replicated the molecular profiles as well as their prognostic value in the replication series of 77 additional independent samples.
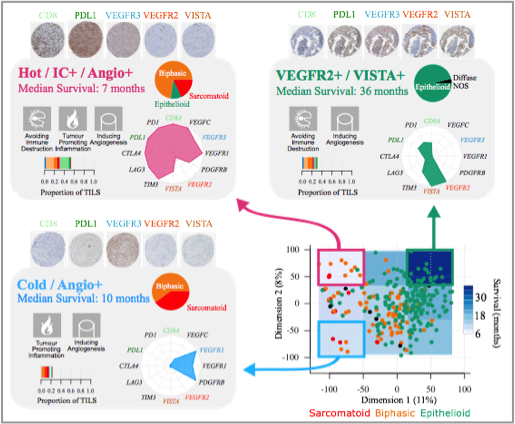
Characteristics of the three MPM transcriptomic profiles. The schematic position of samples harbouring a given profile in the two-dimensional summary (n=284) is represented in the bottom right panel. For each profile, the hallmarks of cancer generally upregulated are indicated by pictograms in the upper left part, the histological type composition is represented by a pie chart in the upper right part, the proportion of tumour infiltrating lymphocytes estimated from the RNA-seq data is represented by a bar plot in the bottom left part, and the expression of representative genes is represented by a radar plot in the bottom right part. Tissue MicroArray IHC staining from the technical validation series, with 0.6 mm core diameter at 5.2× magnification, for the five-gene panel, are presented above each panel (Alcala et al. EBioMedicine 2019).
Through the largest series of whole-genome sequencing data to date, integrated with transcriptomic and epigenomic data using multi-omic factor analysis, we demonstrate that the current WHO classification only accounts for 9% of inter-patient molecular differences. Instead, we propose a novel morpho-molecular classification of MPM based on four dimensions: ploidy, tumor cell morphology, adaptive immune response, and CpG island methylator profile. We show that these four dimensions are complementary, capture major inter-patient molecular differences, and are delimited by extreme phenotypes that, in the case of the interdependent tumor cell morphology and adapted immune response, reflect tumor specialization. These findings unearth the interplay between MPM functional biology and its genomic history, and provide insights into the variations observed in the clinical behavior of MPM patients.
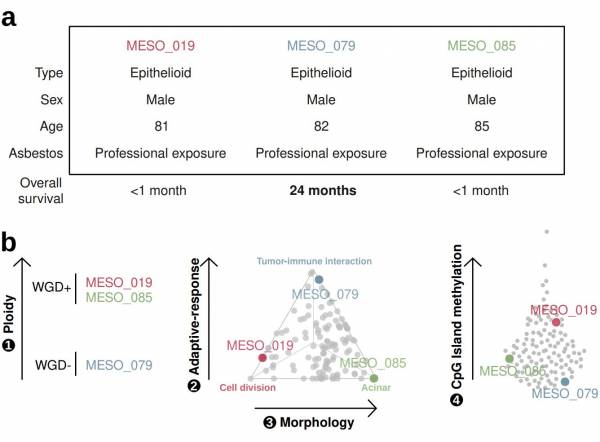
The utility of a four-criteria classification of mesothelioma. Three patients with mesothelioma had similar clinical characteristics yet different outcomes. The three patients have vastly different tumour profiles based on our four-criteria classification. Arrows are directed from low to high values for each criterium, and gray dots represent mesothelioma tumours. (Mangiante et al. Nat Genet 2023).
Our data suggest that MPMs show remarkable morphological intra-tumoral heterogeneity (ITH), both at the cellular composition and the microenvironment, and this has been little studied beyond the histological aspect. Even less is known about how the morphological ITH translates into molecular ITH, despite this might explain the diversity in the response to immunotherapy and anti-angiogenic drugs. We have shown previously that the prognosis of MPM is best explained by a continuum of molecular profiles, with strong differences in tumor immune microenvironment across samples, and that these immune gradients are associated with specific molecular features of the tumor. Studying these interactions is becoming possible with the emergence of single-cell sequencing technologies, which allow to characterize the different cell populations within a tumor, rather than simply the average signal with traditional bulk sequencing. The MESOMICS2 project aims to: (1) build a comprehensive reference molecular map of MPM at the single-cell resolution, (2) quantify and characterize the amount of molecular ITH; and (3) identify predictive candidate biomarkers for aggressiveness and survival that will eventually also guide the design of future clinical trials. Altogether this research will provide insight in biological mechanisms by disentangling the intricate convolution of the signal from the microenvironment with that of the tumor that could not be resolved until now. This will result in biomarkers with stronger diagnostic, prognostic, and predictive values.
FUNDING
Understanding the spatiotemporal eco-evolutionary interactions in malignant pleural mesothelioma. Worldwide Cancer Research (WCR, UK). Grant 24-0106. Coordinators. Active
Intra-tumour heterogeneity of pleural mesothelioma at the single cell level. Department of Defence (DoD, USA). Coordinators. Active.
Tremplin – ERC Consolidator Grant (T-ERC CoG). Agence National de la Recherche (ANR, France). Coordinators. Active
Transcriptomic characterization of biphasic and sarcomatoid malignant pleural mesotheliomas. La Ligue Contre le Cancer (LNCC, France). Appel d’offres 2020. Coordinators. Completed.
Unveiling the intra-tumour heterogeneity of malignant mesothelioma. La Ligue Contre le Cancer (LNCC, France). Appel d'offres 2017. Coordinators. Completed.
Molecular characterisation of malignant pleural mesothelioma. Institut National Du Cancer (INCa, France). PRT-K16-039. Coordinators. Completed.


RATIONALE
In the past years, advancements in artificial intelligence (AI) have had a significant influence on many aspects of our lives. It is especially tempting to use AI for the study of histopathological tissue sections, which requires highly trained pathologists to evaluate pictures with a gigapixel resolution or more. In the context of rare cancers, the rarity of cases may lead to diagnosis uncertainty, delay, or even misdiagnosis. AI has the potential to make diagnostic procedures more effective, repeatable, and reliable.
The aim of this transversal project is to translate our multi-omics tumor profiling into the clinical setting without the need to generate costly and complex-to-analyze molecular data. To do this, we are exploring how different deep-learning computer vision algorithms can detect morphological features that pathologists will be able to recognize, with the ultimate goal to improve the diagnosis and treatment of rare cancers.
AIMS AND METHODS
Anomaly detection deep learning algorithms are a type of unsupervised machine learning algorithm that are used to identify patterns in images that are unusual or unexpected. They do not require any prior knowledge of what an anomaly is, and simply need to be trained on “normal” images. This “normal” class can be defined in different ways, e.g. good prognosis patients, a specific molecular group etc. allowing for the identification of discriminating regions for tumor proliferation or aggressiveness (see Figure below).
We are also developing deep learning algorithms to detect mitosis and high proliferation areas based on immunohistochemistry whole slide images. Computer vision algorithms on whole slide images (WSI) used together with molecular data can uncover morphological features with the potential to reconcile discordant molecular and histopathological classifications.
This technology has the potential to simplify and enrich clinical decision-making processes, but requires rigorous external validation in clinical settings. For this purpose, the Rare Cancers Genomics initiative maintains a rich network of collaborators (ENETS, EPIC, MESOBANK, EURACAN).
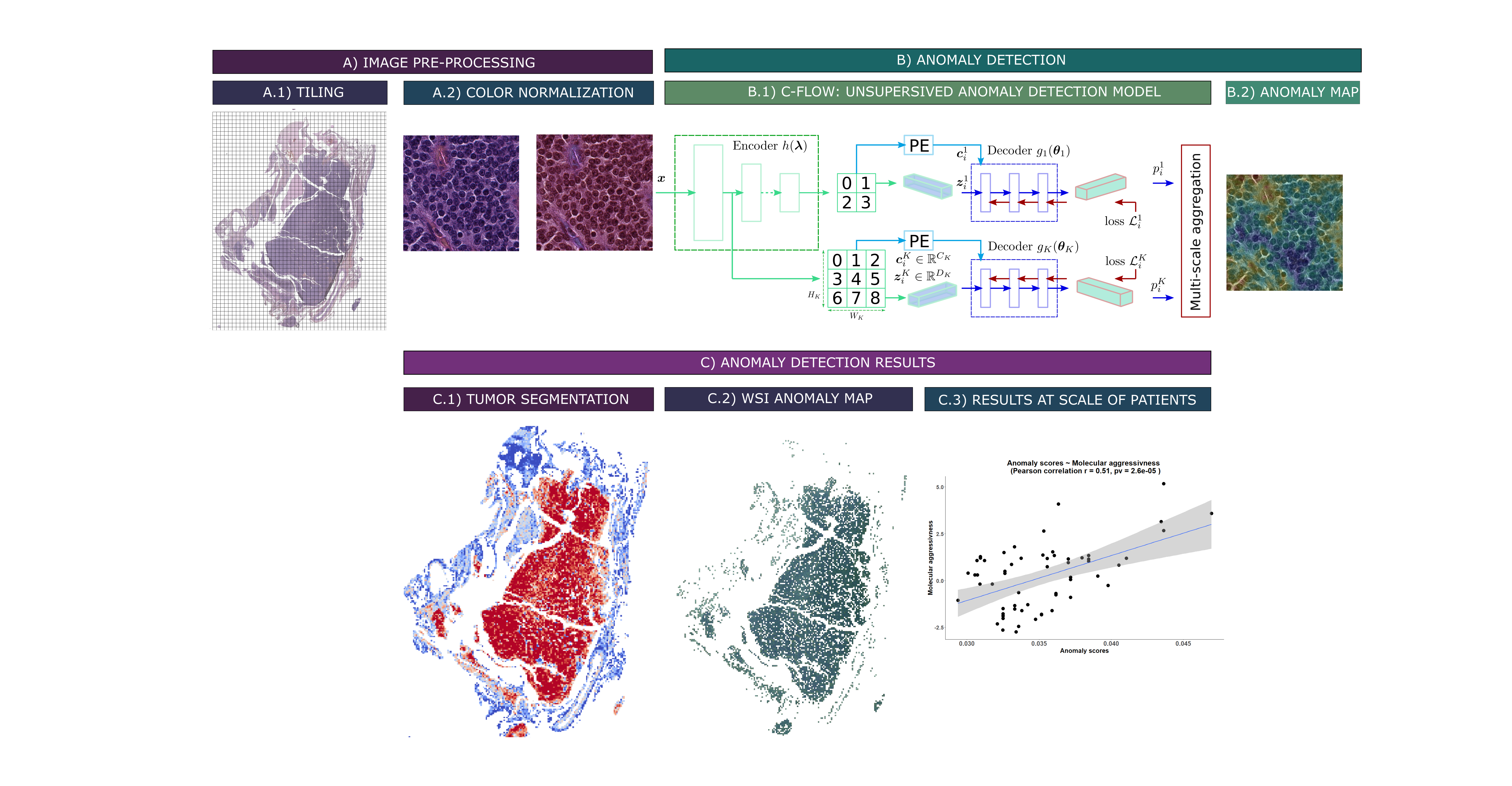
Unsupervised anomaly detection pipeline for whole-slide images of lung carcinoids (WSI): A) Preprocessing steps. A.1) Tiling of the WSI. A.2) Vahadane color normalization (Vahadane et al. 2016). B) Deep learning method for anomaly detection. B.1) Unsupervised anomaly detection model C-Flow via normalization flow (Gudovskiy et al. 2022). B.2) Pixel-level tiled anomaly map. C) Preliminary results. C.1) Segmentation of WSI tumors, via a C-Flow model trained on tumor images. The bluer a tile is, the more it falls outside the learned distribution, and thus the more it is non-tumoral. C.2) Map of WSI abnormalities at the pixel scale, obtained via a model trained on typical carcinoid (TC) samples. The more abnormal a pixel is, i.e. reddish, the more it denotes a deviation from the TC distribution. C.3) Aggregate abnormality score by patients, correlated to their “molecular aggressiveness” defined using multi-omics data.
VIDEOS


RATIONALE
Cancer is a disease of the genome, governed by principles of Darwinian evolution, whereby a cell acquires successive genetic or epigenetic alterations that lead to malignant transformation. The microenvironment is responsible for the selective pressures acting upon cells, for example immune predation, or resource availability (e.g. oxygen and nutrients provided through vascularization), and can in turn be remodeled by the cancer cell. Ecological interactions between a tumor and its environment are thus increasingly scrutinized to understand carcinogenesis and tumor progression.
AIMS
The cancer ecology and evolution project of the rare cancers genomics team is a transversal research program led by Dr. Nicolas Alcala that aims to build a theoretical and analytical framework of cancer formation and development. The project makes use of existing mathematical and computational models, as well as development of new methods, applying them to the multi-omic data generated within other team projects (lungNENomics, panNENomics, MESOMICS, and SARCOMICS). We focus on three topics :
- 1. Reconstructing the evolutionary history of tumors, and inferring the evolutionary forces at work, such as mutation and tumor demography (neutral and selection-driven growth). We use genomic and epigenomic data to infer the sequence of events that occurred in the tumor.
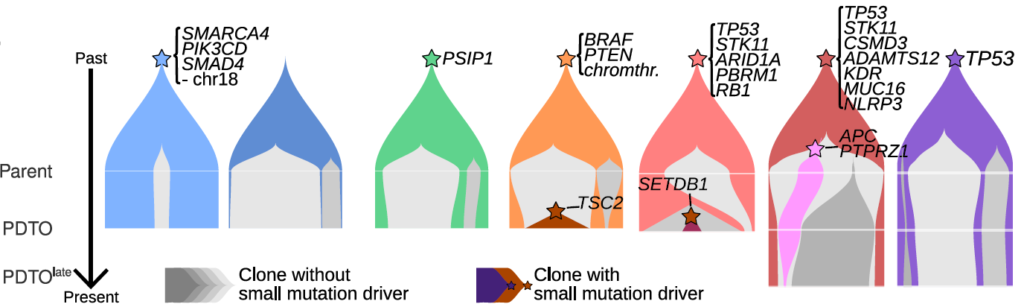
Temporal dynamics of tumor clones (colored shapes) in patient-derived tumor organoids (PDTO) of neuroendocrine neoplasms. Clonal complexity and selection (gray: neutrally evolving clones; colors: selected clones) increase with grade (left: low grade; right: high grade). From Dayton, Alcala et al. (Under review) https://www.biorxiv.org/content/10.1101/2022.10.31.514549v1
- 2. Understanding the interaction between tumor and microenvironment, and how they shape the selective landscape of tumors and their evolutionary trajectories. We couple transcriptomic, genomic, and histopathological data to infer tumor phenotypes, create a genotype-phenotype map and link it with the tumor microenvironment.
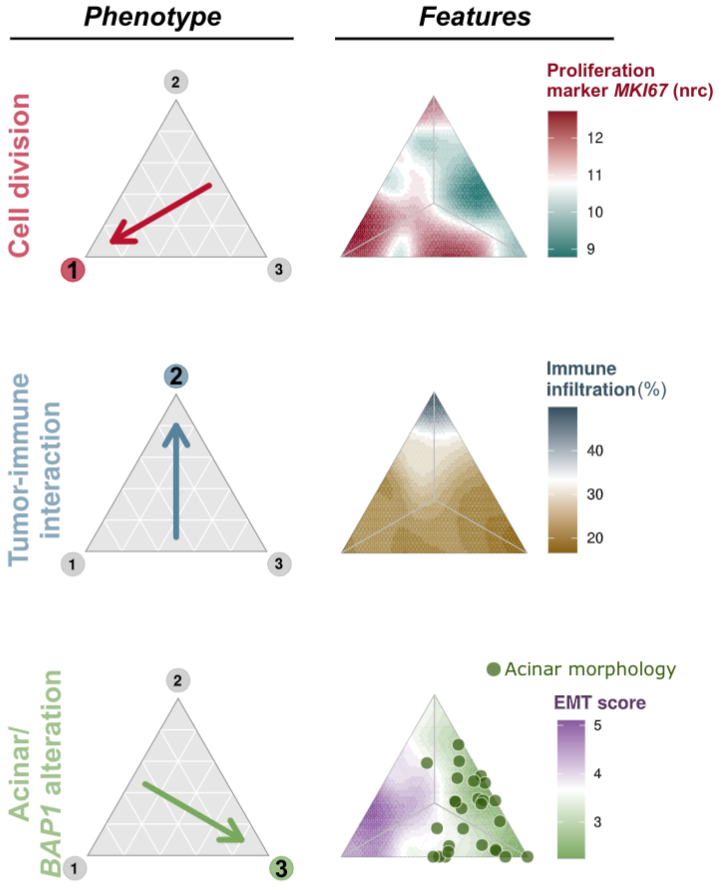
Malignant pleural mesothelioma phenotypes and associated features. Mesothelioma can be classified into three phenotypes (cell division, tumor-immune interaction, acinar), and each evolved under specific environmental conditions. From Mangiante, Alcala, Sexton-Oates, Di Genova et al., Nature Genetics (In press) https://www.biorxiv.org/content/10.1101/2021.09.27.461908v1
- 3. Developing statistical tools to quantify cell population evolvability, with the aim to anticipate malignant transformation, increased aggressiveness, and resistance to treatment. We use mathematical models to predict the behavior of certain quantities describing the intra-tumor heterogeneity of a population and mathematical statistics to design the best tool.
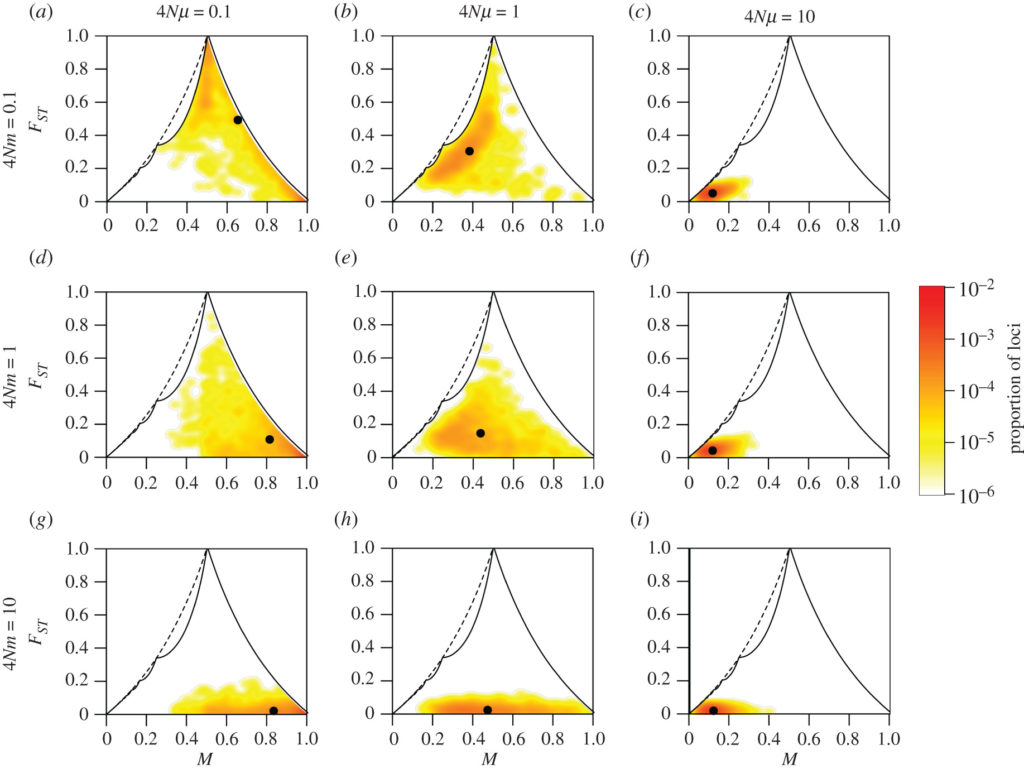
Computer simulations of a measure of differences in genetic composition (ranging from 0 for genetically similar populations to 1 for completely different populations), as a function of the frequency of genes M in two populations. Rows correspond to different rates of migration between the two populations (4Nm) and columns to different mutation rates for the individuals (4Nmu). From Alcala and Rosenberg, Philosophical Transactions of the Royal Society B https://royalsocietypublishing.org/doi/full/10.1098/rstb.2020.0414
FUNDING
Understanding the spatiotemporal eco-evolutionary interactions in malignant pleural mesothelioma. Worldwide Cancer Research (WCR, UK). Grant 24-0106. Coordinators. Active
Reconstructing the evolutionary history of neuroendocrine tumor subtypes. Neuroendocrine Tumor Research Foundation (NETRF, US). 2023 Mentored Award. Coordinators. Active
PhD fellowship to Laurane Mange. La Ligue contre le cancer. 2023-2026. Active
Population genetics of cancer evolution. France-Stanford Center for Interdisciplinary Studies (US). 2020-2021 Collaborative Research Project Grant. Active